(This article is the outcome of a project collaboration between the Tang Prize Foundation and The Investigator Taiwan.)
The Chinese original by Ching-Ting Tsai
Reviewed by Yan-An Chang
The 2018 Tang Prize in Biopharmaceutical Science was jointly awarded to Dr. Tony Hunter, Dr. Brian Druker and Dr. John Mendelsohn “for the discovery of protein tyrosine phosphorylation and tyrosine kinases as oncogenes, leading to successful targeted cancer therapies,” as the citation reads.
In living organisms, proteins often become activated after they are phosphorylated, which in turn triggers the signal transduction carried out by downstream molecules. The enzyme that catalyzes the process of phosphorylation is called kinase. As protein phosphorylation plays a key role in regulating many cellular activities, if mutations occur on these kinases, cells will overpopulate and then cancer occurs. In this kinase family, the receptor tyrosine kinases (RTK) are transmembrane proteins. They are closely associated with cell growth because they “bind and respond to many growth factors.”[i] Thus, the overexpression of RTKs was found to be highly correlated with cancer.
Tyrosine kinase inhibitors (TKIs) came to be a very effective tool to curb the over proliferation of cells found in many diseases. This kind of treatment, known as targeted cancer therapy, has enjoyed tremendous success especially in tackling chronic myelogenous leukemia (CML). Dr. Brian Druker, one of the 2018 laureates, made a great contribution in this field by developing imatinib (Gleevec®), a drug that is aimed at CML. It fundamentally changed the clinical approach to CML and improved the quality of life for patients who in the past had to wait for a matching donor in order to undergo a marrow transplant.
95% of the time, CML is caused by an abnormality observed in chromosome 9 and 22. When part of chromosome 9 and part of chromosome 22 break off and switch places, the Abl gene on chromosome 9 is translocated and joins to the breakpoint cluster region (BCR) gene on chromosome 22, producing a shortened chromosome 22 which encodes the Bcr-Abl fusion gene. This gene is then transcribed and translated into a hybrid Bcr-Abl fusion protein “with dysregulated (significantly enhanced) tyrosine kinase activity,” resulting in “the aberrant growth and differentiation of leukemic cells in CML”[ii] In the bone marrow of patients with CML, a group of white blood cells called granulocytes will grow uncontrollably and the increased number of granulocytes in the blood will bring about symptoms such as the enlargement of both the liver and spleen or abnormal bleeding.
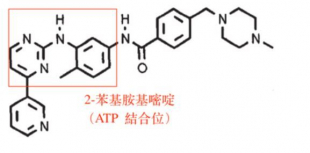
Tyrosine phosphorylation takes place when a kinase removes a phosphate group from ATP and attaches it to an amino acid of a substrate protein. Therefore, when developing a drug to inhibit the activities of tyrosine kinases, many scientists tend to take a more conventional approach and focus on the ATP binding site where this process begins. However, the compounds thus synthesized didn’t exhibit high specificity, and would inhibited other kinds of kinases as well. The main goal therefore was to develop a selective inhibitor that can distinguish one kinase from another. To resolve this problem, Dr. Druker and his team synthesized “a series of compounds of the 2-phenylaminopyrimidine class” that was already identified by other scientists and screened them “for the ability to inhibit a panel of protein kinases.” Eventually, “CGP 57148 (Fig. I) was found to be a potent inhibitor of the Abl protein tyrosine kinase” since “inhibitors of this class act as competitive inhibitors of protein kinases with respect to ATP”[iii] CGP 57148 is the predecessor of imatinib. This compound is so effective because it not only fits into the active site where ATP would normally bind with a tyrosine kinase, but it can also change the conformation of a tyrosine kinase by inducing its active site to refold to an inactive shape so that it will no longer be able bind with ATP or its substrate. (Fig. II) The clinical trials of imatinib were huge successes too, and patients experienced only mild adverse effects. In the case of another TKI, cetuximab, while it “induce(s) EGFR (epidermal growth factor receptor) internalization, most receptors subsequently translocated into the late endosome, leading to lysosomal degradation.”[iv] Dr. Druker and his team should be credited with coming up with a very effective therapy to switch off the mechanism of tyrosine phosphorylation that causes cancer cells to grow. Besides CML, imatinib has also been used in patients with gastrointestinal stromal tumor (GIST), mastocytosis, and myelodysplastic syndromes, as the mechanisms underlying all these three diseases are similar to that of CML. In addition, he and his team have participated in the development of antibodies that can identify and characterize tyrosine kinase substrates and have also built up a comprehensive system to evaluate potential tyrosine kinase inhibitors.
TKIs are the foundation for the development of several cancer therapies. They revolutionized the treatment of many diseases caused by abnormal cell proliferation. From the understanding of the mechanisms that drive the growth of cancer, to the development of a specifically targeted agent, to the implementation of clinical trials, the research conducted in Dr. Druker’s laboratory not only demonstrates the whole process of the development of TKI drugs, but also helps us gain a deeper knowledge of the pathology of cancer. Scientists are hopeful that in the future, this valuable information will be used as effective methods for combating terrible diseases such as cancer.
[i] “RTK.”
[ii] “BCR-ABL RNA PCR Quantitation for Leukemia.”
[iii] “Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells,” p.561
[iv] “EGFR Downregulation after Anti-EGFR Therapy Predicts the Antitumor Effect in Colorectal Cancer.”
References:
1.Department of Hematological Malignancies, Knight Diagnostic Laboratories, OHSU. “BCR-ABL RNA PCR Quantitation for Leukemia.” https://knightdxlabs.ohsu.edu/home/test-details?id=BCR-ABL+RNA+PCR+Quantitation+for+Leukemia
2.Druker, B. J., Tamura, S., Buchdunger, E., Ohno, S., Segal, G. M., Fanning, S., & Lydon, N. B. (1996). “Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr–Abl positive cells.” Nature medicine, 2(5), p.561
3.Okada Y, Kimura T, Nakagawa T, Okamoto K, Fukuya A, Goji T, Fujimoto S, Sogabe M, Miyamoto H, Muguruma N, Tsuji Y, Okahisa T, Takayama T.(2017) “EGFR Downregulation after Anti-EGFR Therapy Predicts the Antitumor Effect in Colorectal Cancer.” Mol Cancer Res. 2017 Oct;15(10):1445-1454. doi: 10.1158/1541-7786.MCR-16-0383
4.Scitable By Nature Education. “RTK.” https://www.nature.com/scitable/topicpage/rtk-14050230/